Research
New technological and experimental advances have resulted in the massive accumulation of biological measurements, requiring computational analysis to facilitate data interpretation. I develop algorithms to analyze complex biological datasets that draw from core areas of computer science and mathematics.
Computational Systems Biology
Reconstructing Signaling Pathways
Signaling pathways describe the series of reactions that occur when a cell receives an external stimulus and elicits a downstream transcriptional response. We develop methods to computationally analyze signaling pathways, both from manually-curated databases such as KEGG, Reactome, and NetPath as well as large, experimentally derived networks of protein-protein interactions (called interactomes). In particular, we have developed methods to automatically reconstruct signaling pathways similar to the manually-curated data from the larger and noisier interactomes.
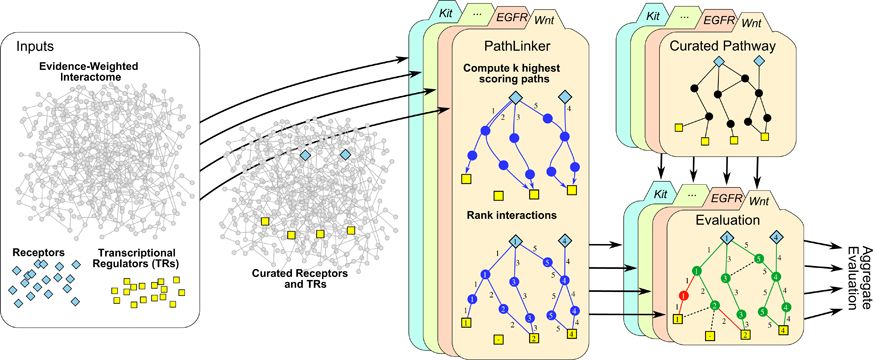
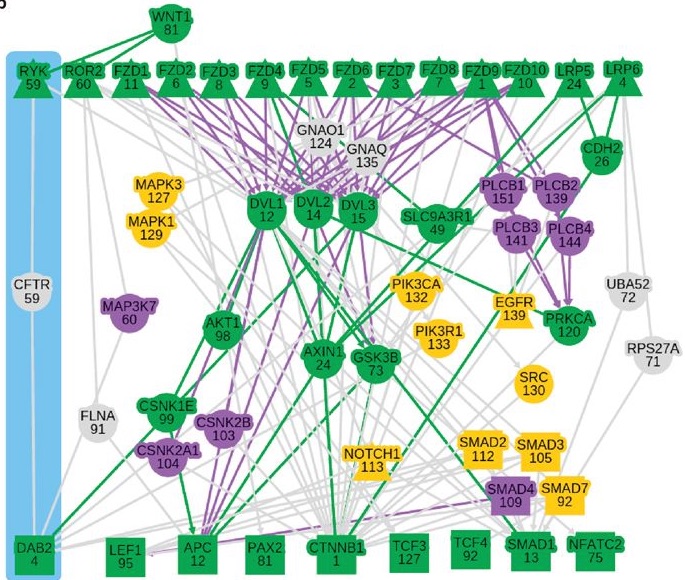

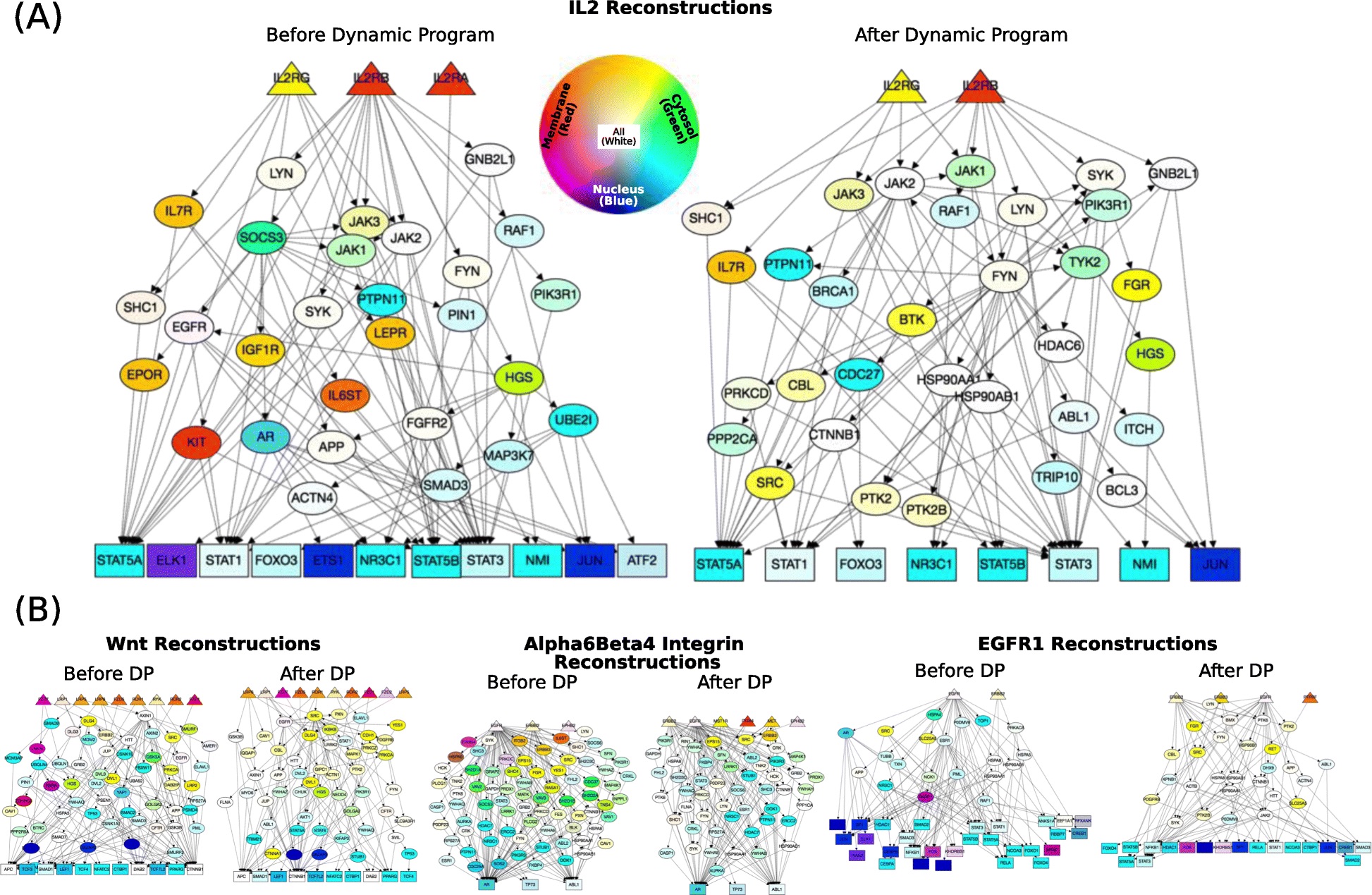
References: Slide 1; Slide 2; Slide 3; Slide 4; Slide 5
Hypergraphs as Alternative Representations of Signaling Pathways
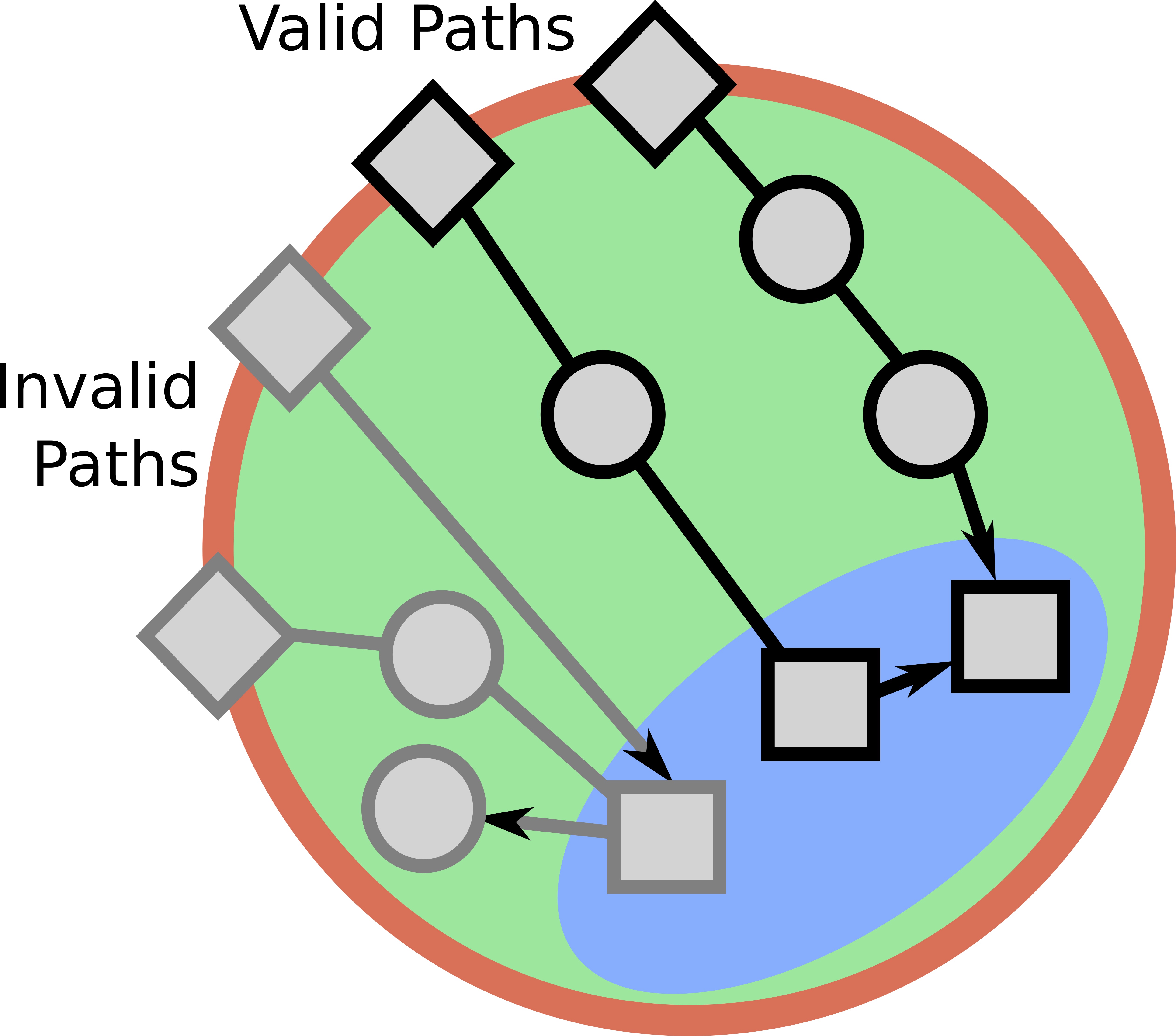
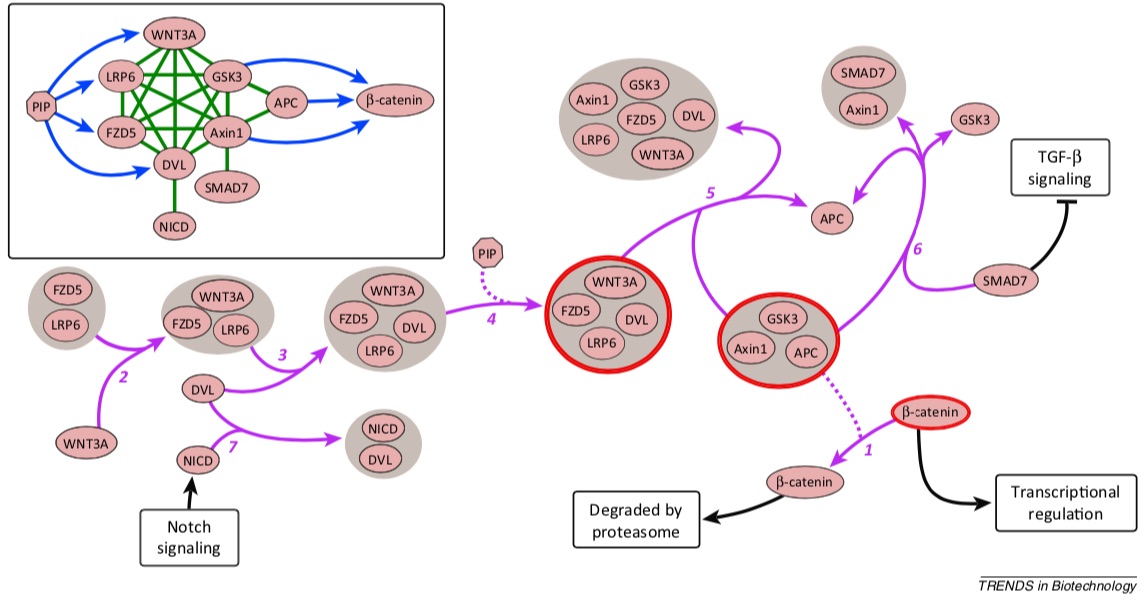
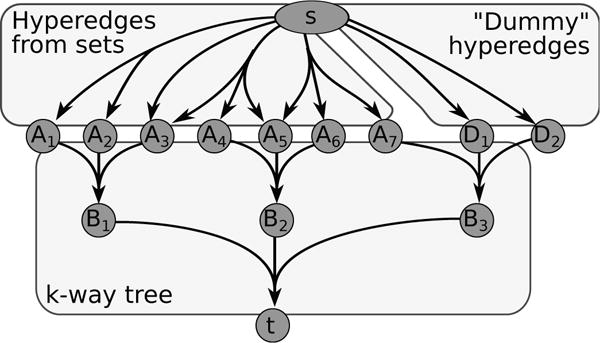
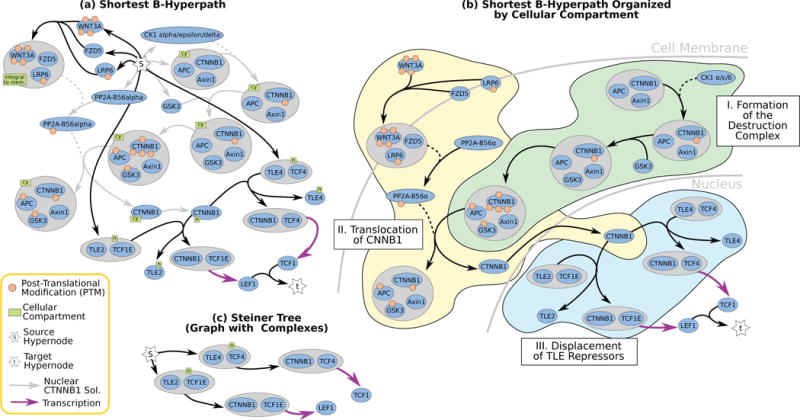
Directed and undirected graphs have been immensly useful for analyzing signaling pathway responses. However, there are known limitations of this representation. For example, multiple proteins or protein subunits may physically join to form protein complexes, which are difficult to represent in graphs due to the pairwise nature of graph edges. For the same reason, the assembly and disassembly of these protein complexes and other regulatory interactions cannot be accurately described using graphs. We are formalizing and designing algorithms for signaling hypergraphs, a reaction-centric representation that better captures the complexity of complex assembly and regulation found in signaling pathways.
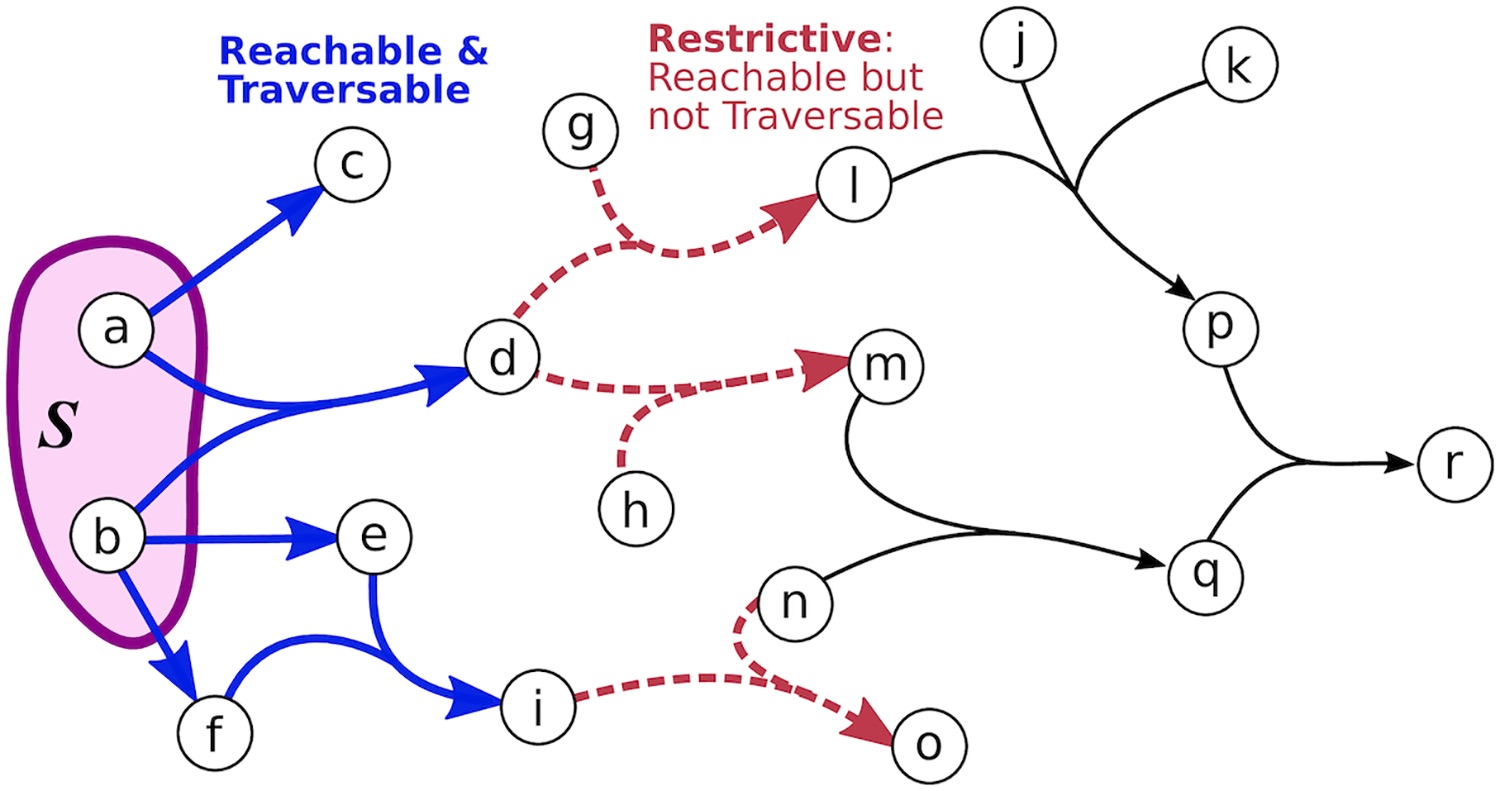
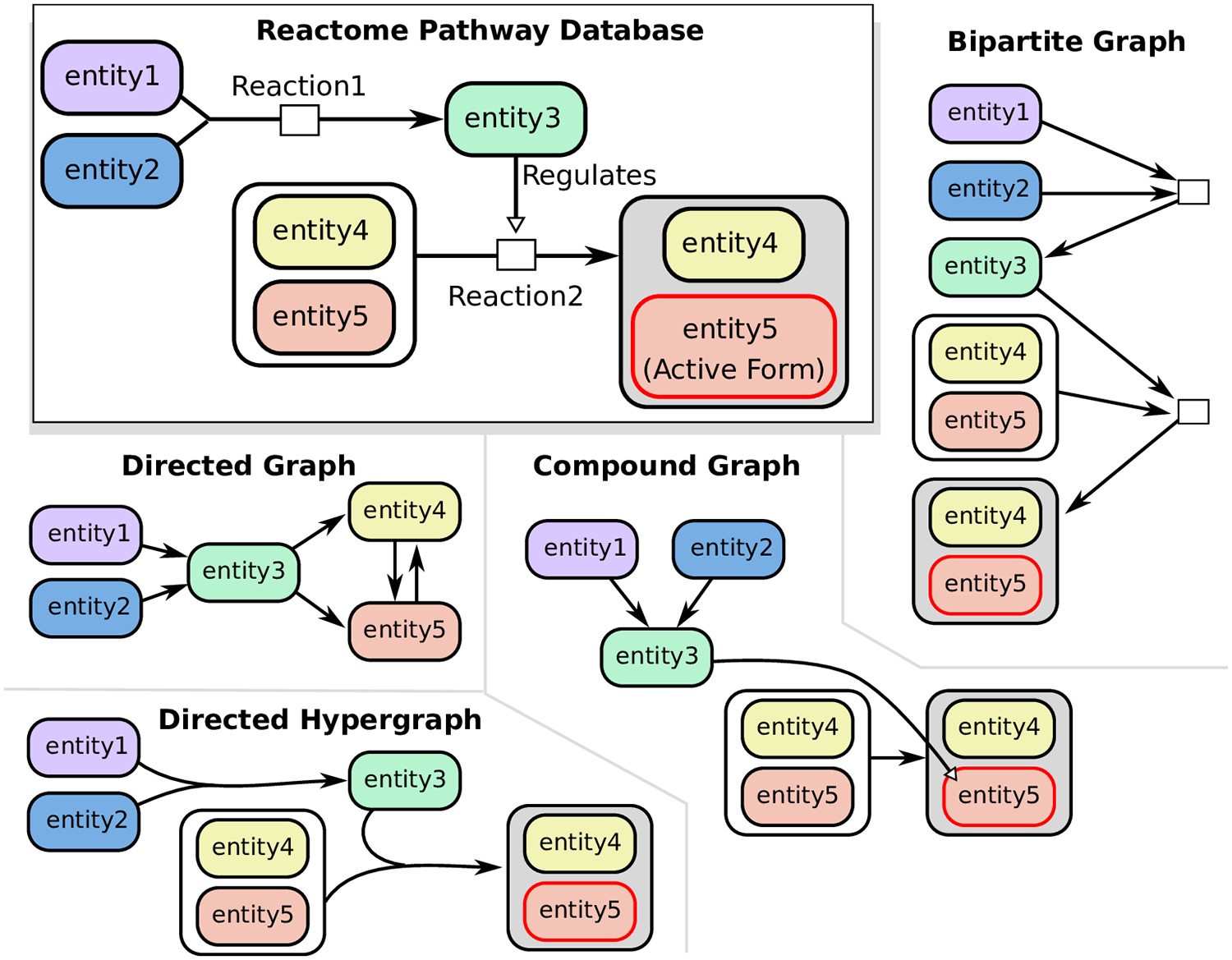
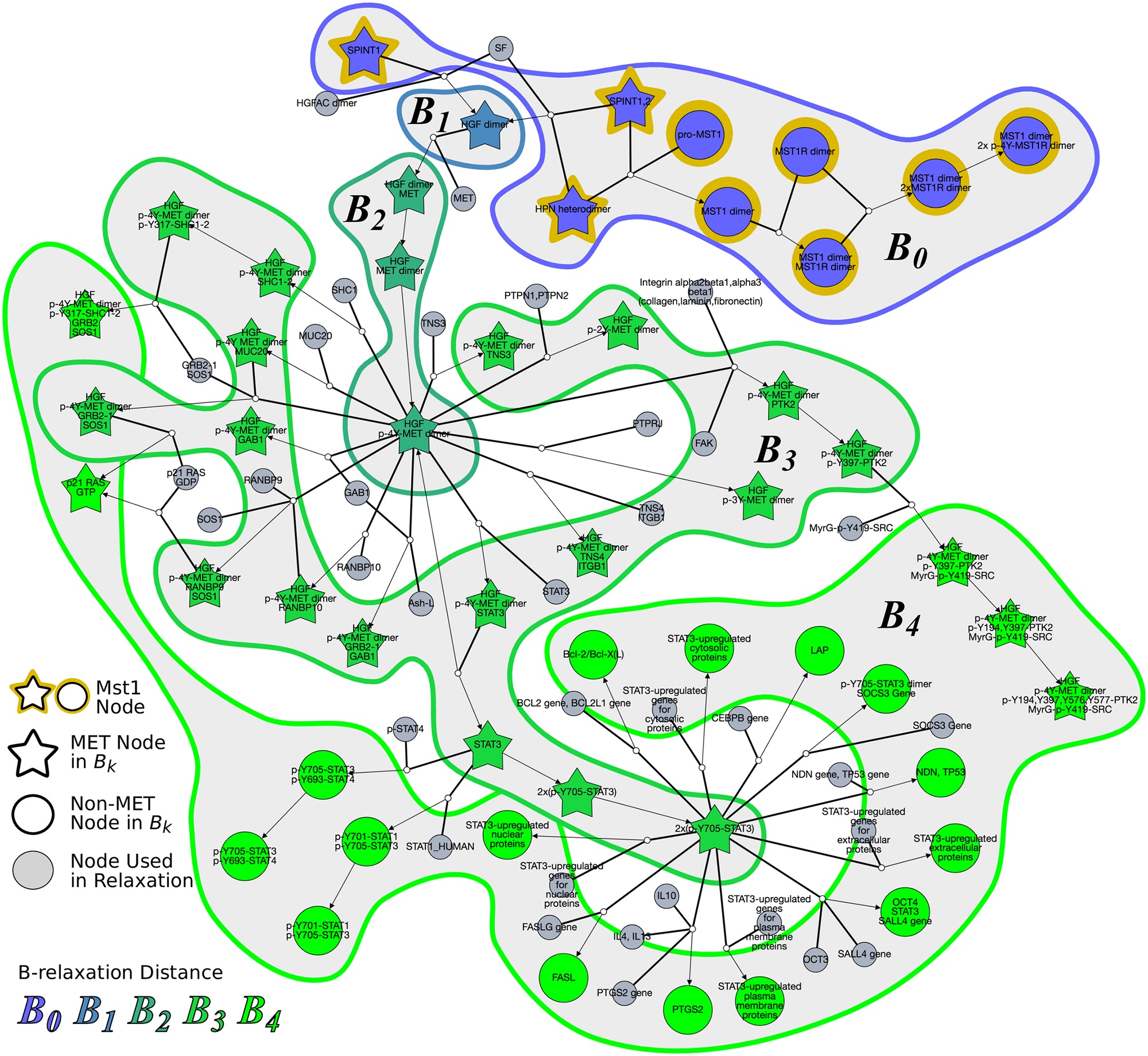
References: Slide 1; Slide 2; Slide 3; Slide 4; Slide 5; Slide 6
Network-Based Disease Gene Prediction
Interactomes and functional interaction networks have been widely used in other computational biology applications. In another research area, we develop algorithms to predict genes that may be associated with a particular disease (e.g. schizophrenia or cancer) given an interactome and a set of known disease genes. We also have worked to identify disease genes that may be associated with a specific disease phenotype, such as aberrant cell motility.
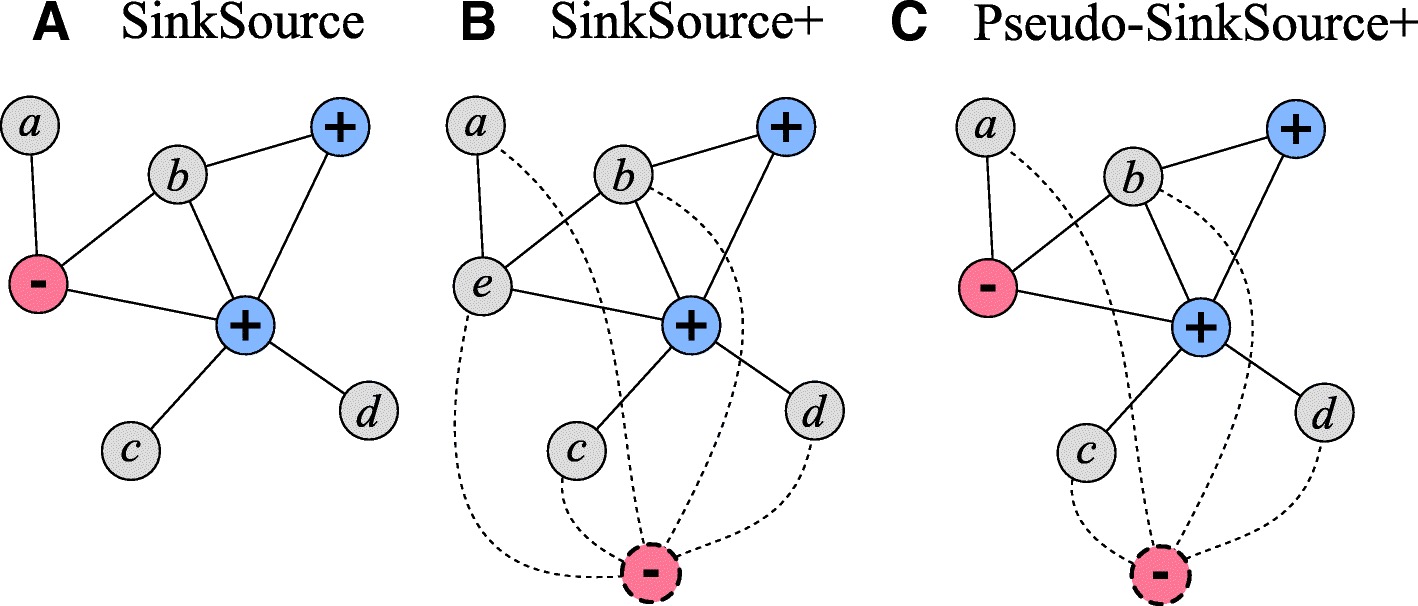
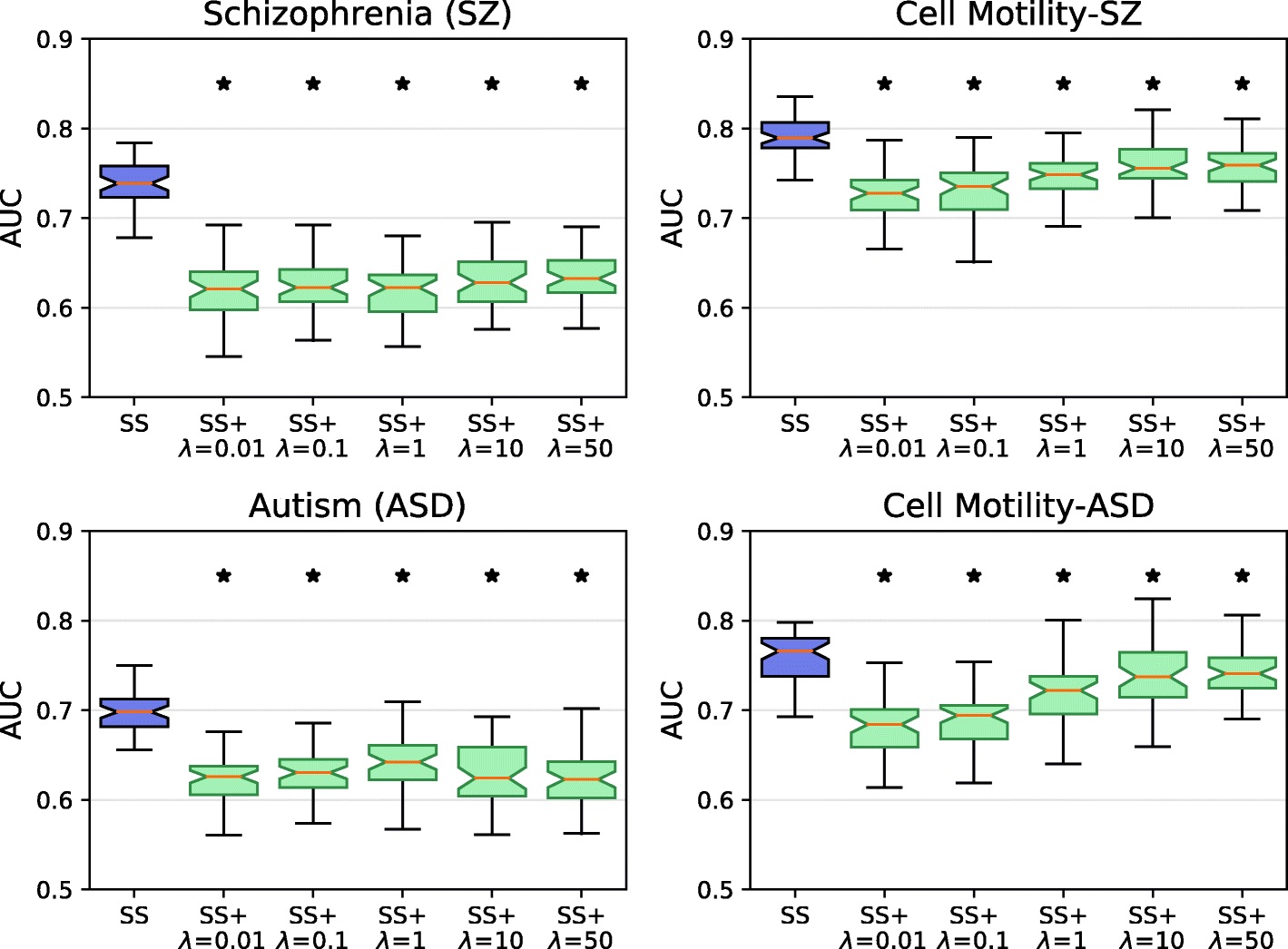
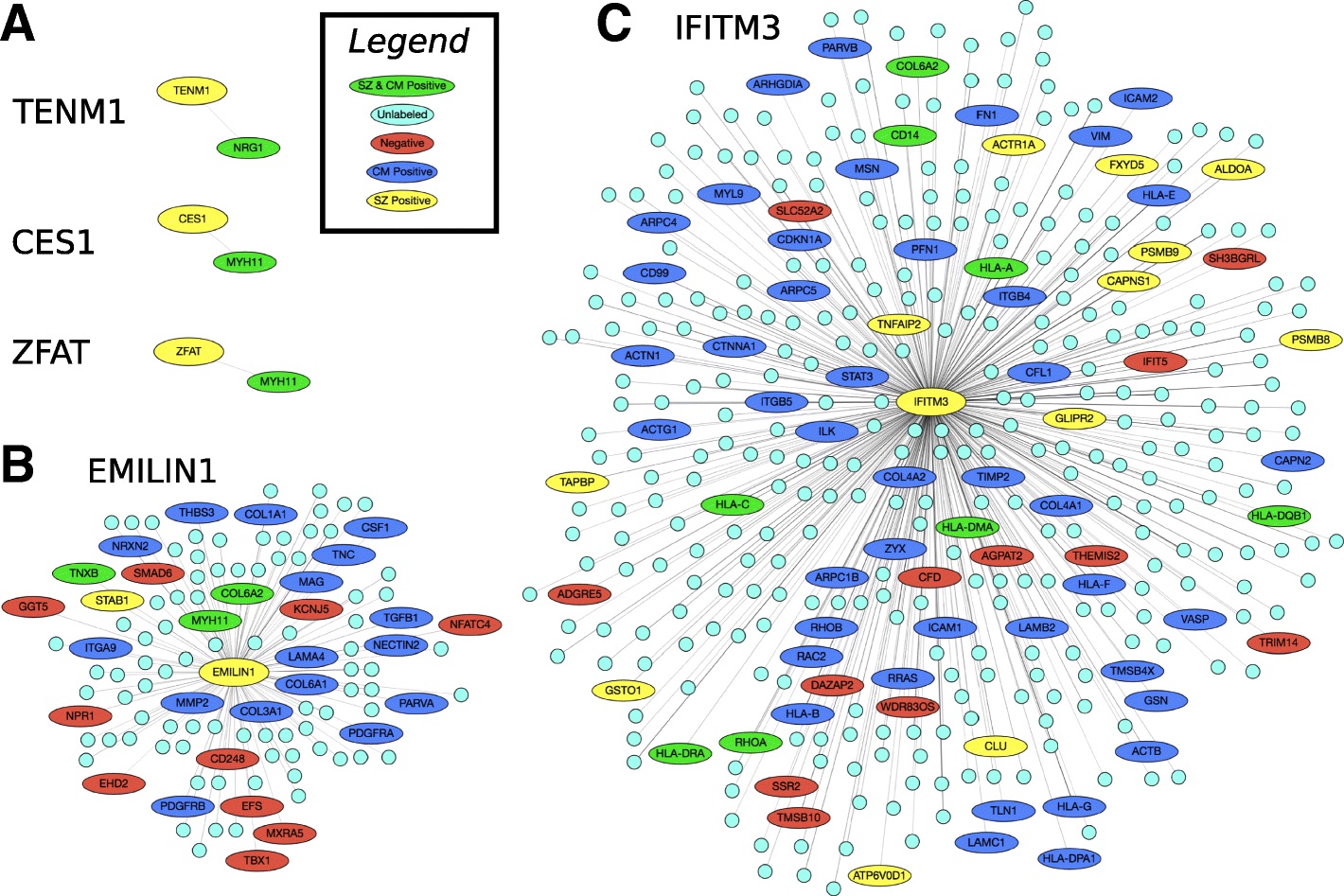
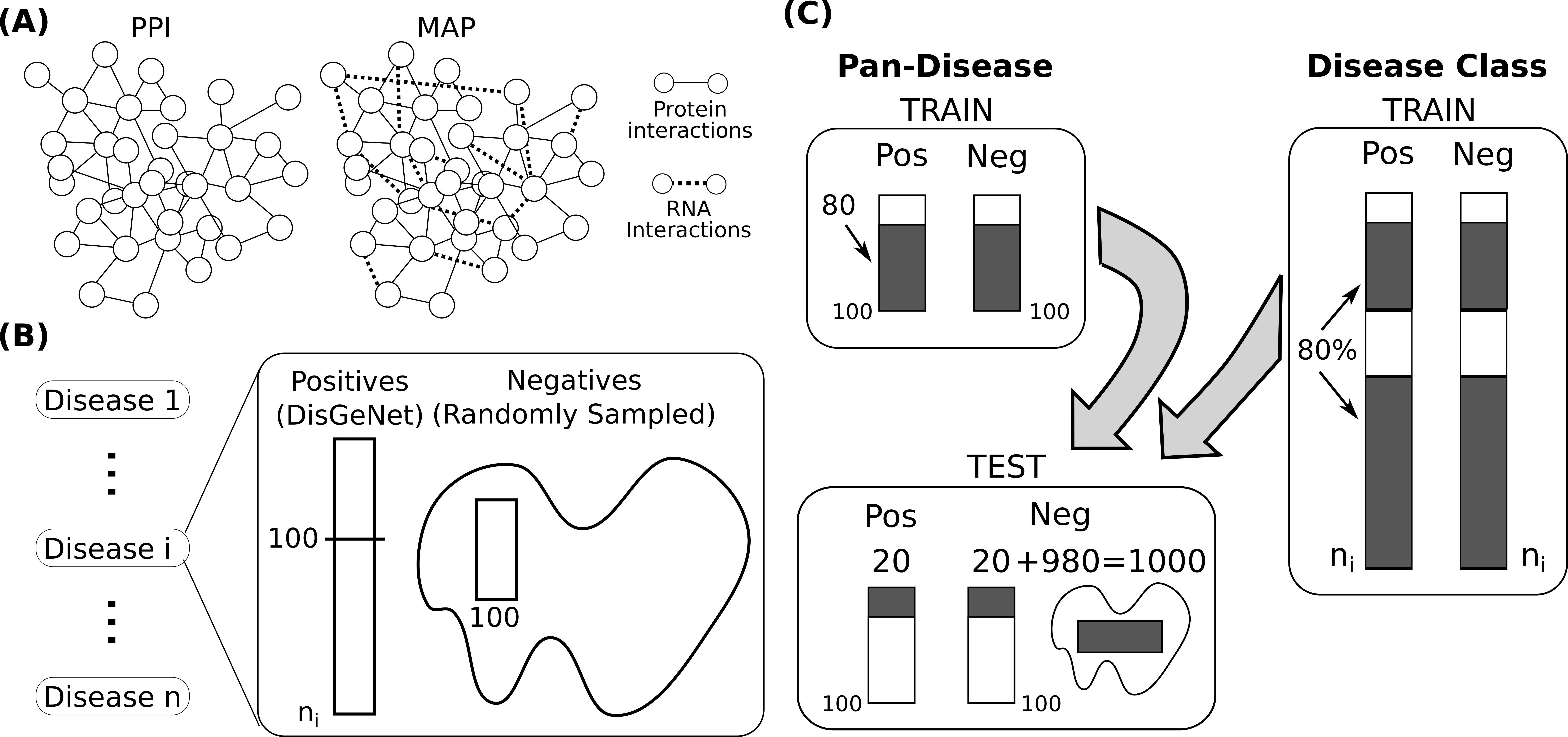
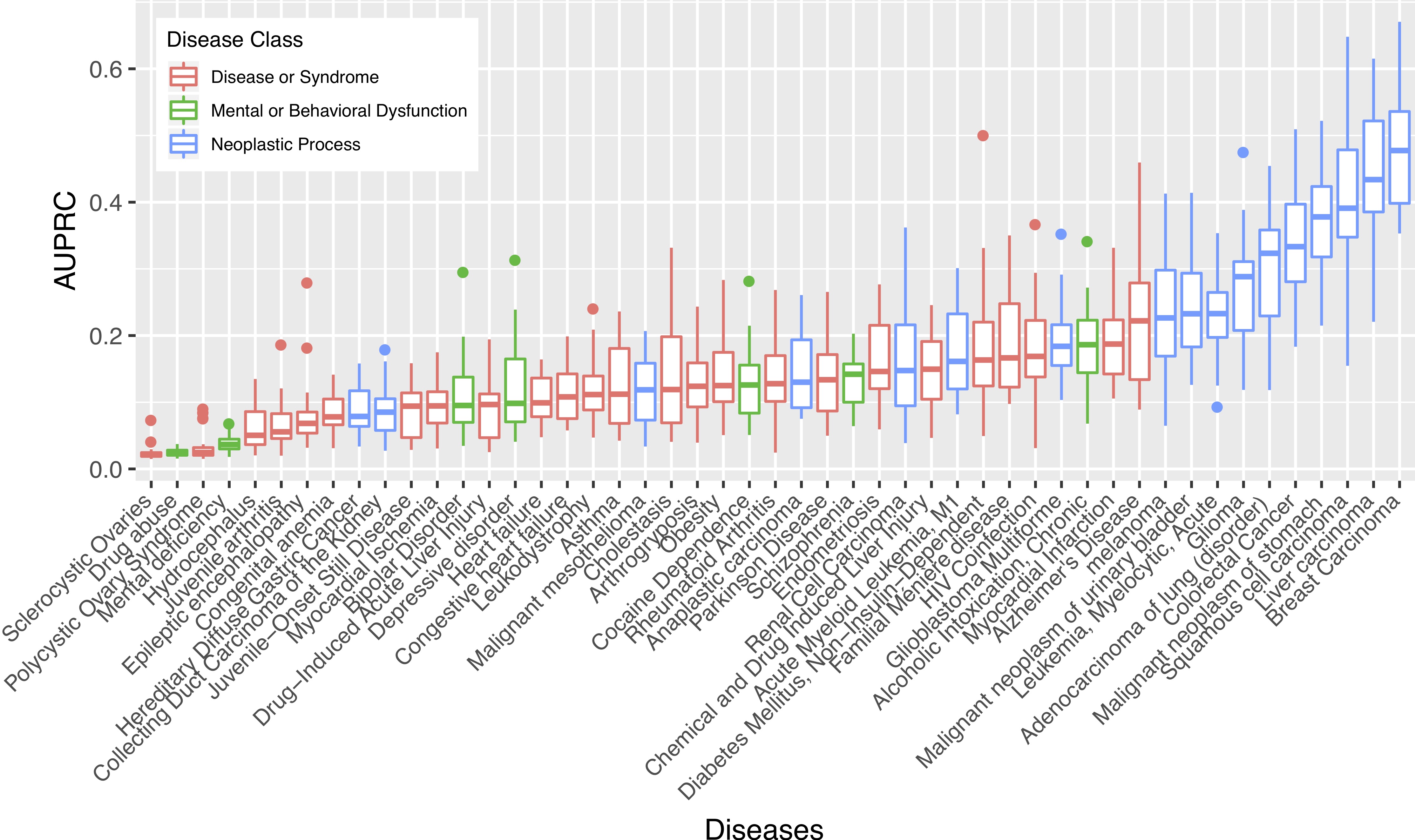
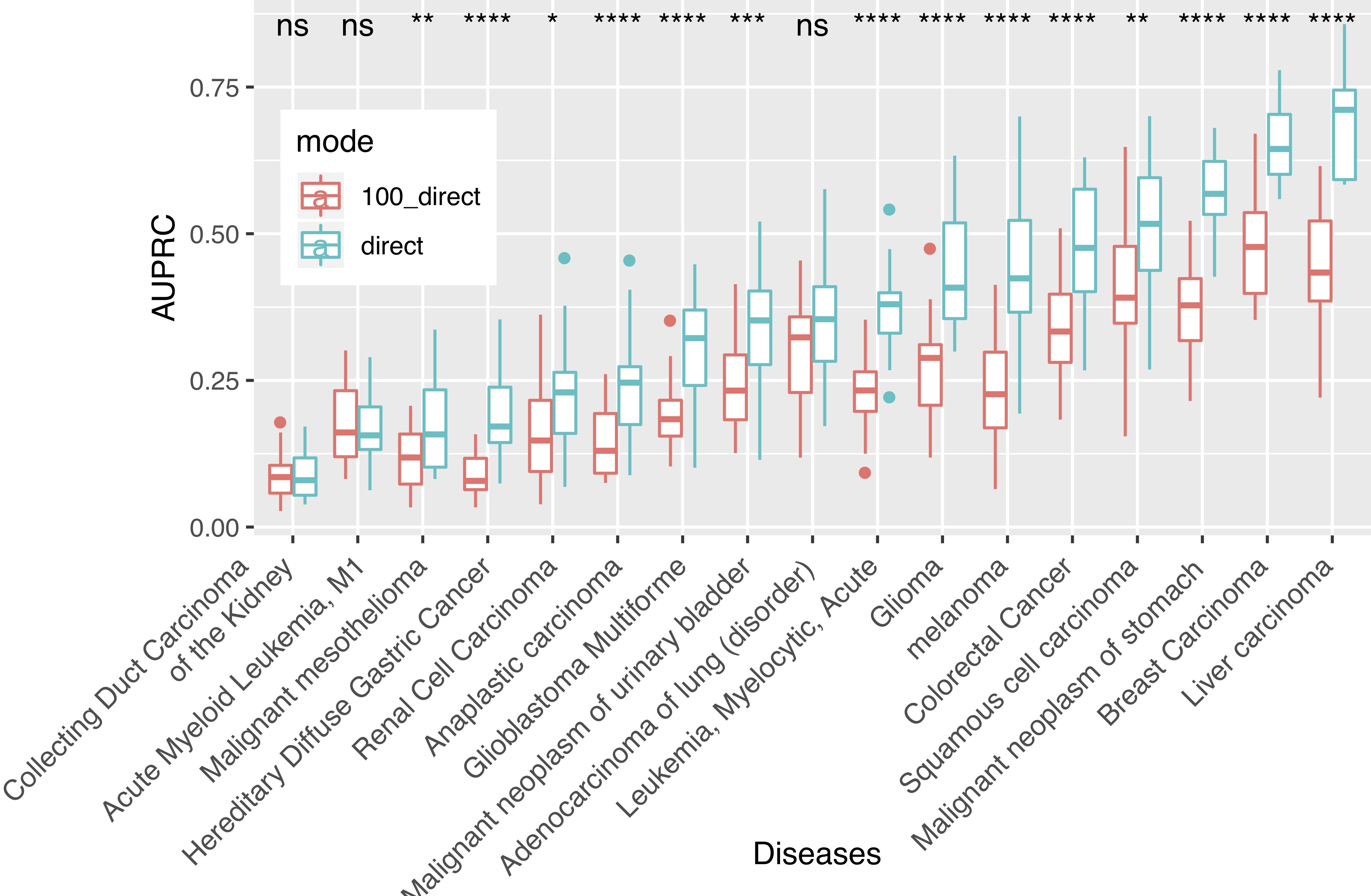
References: Slide 1; Slide 2; Slide 3; Slide 4; Slide 5; Slide 6
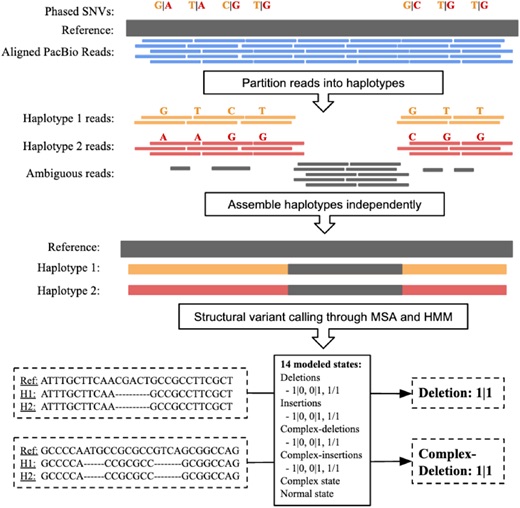
Structural Variant Detection
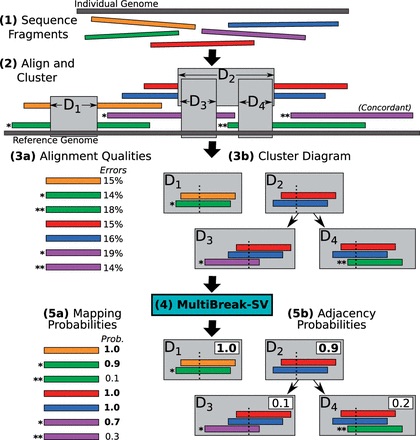
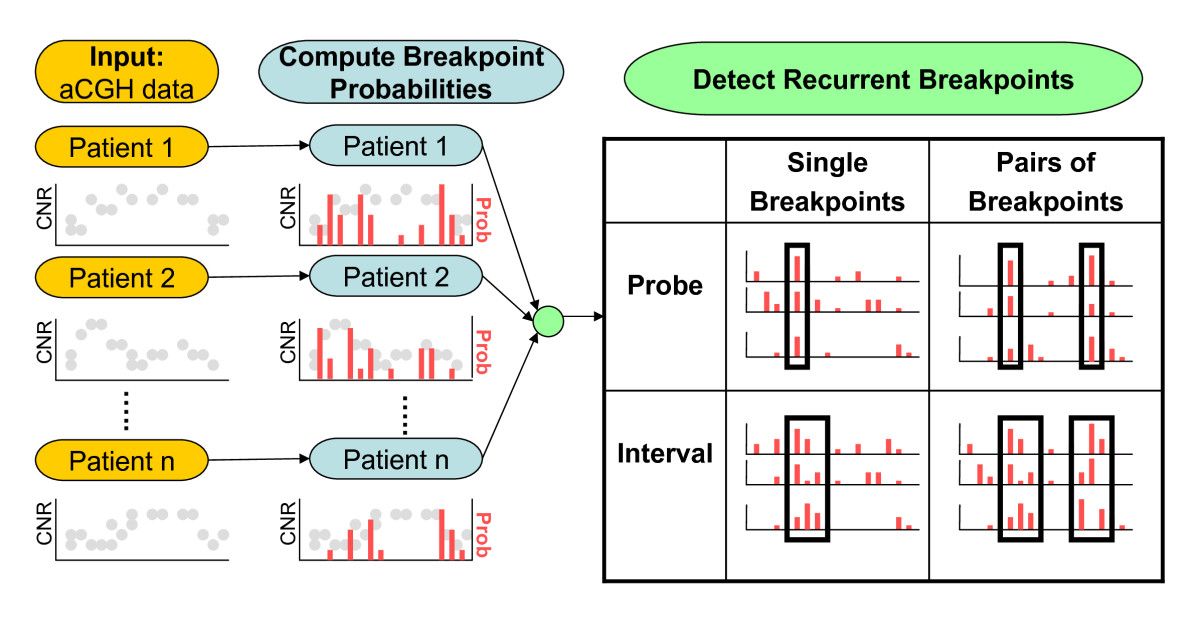
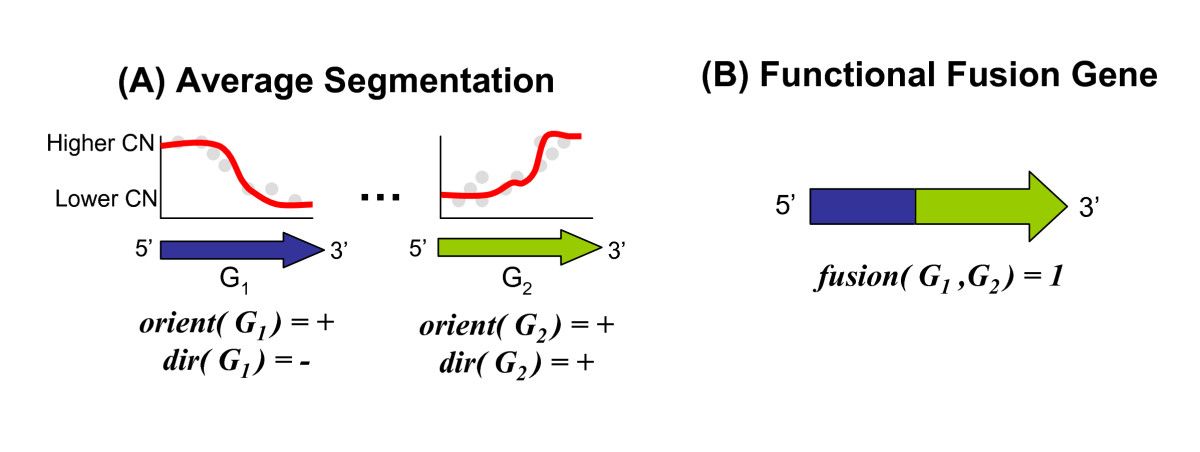
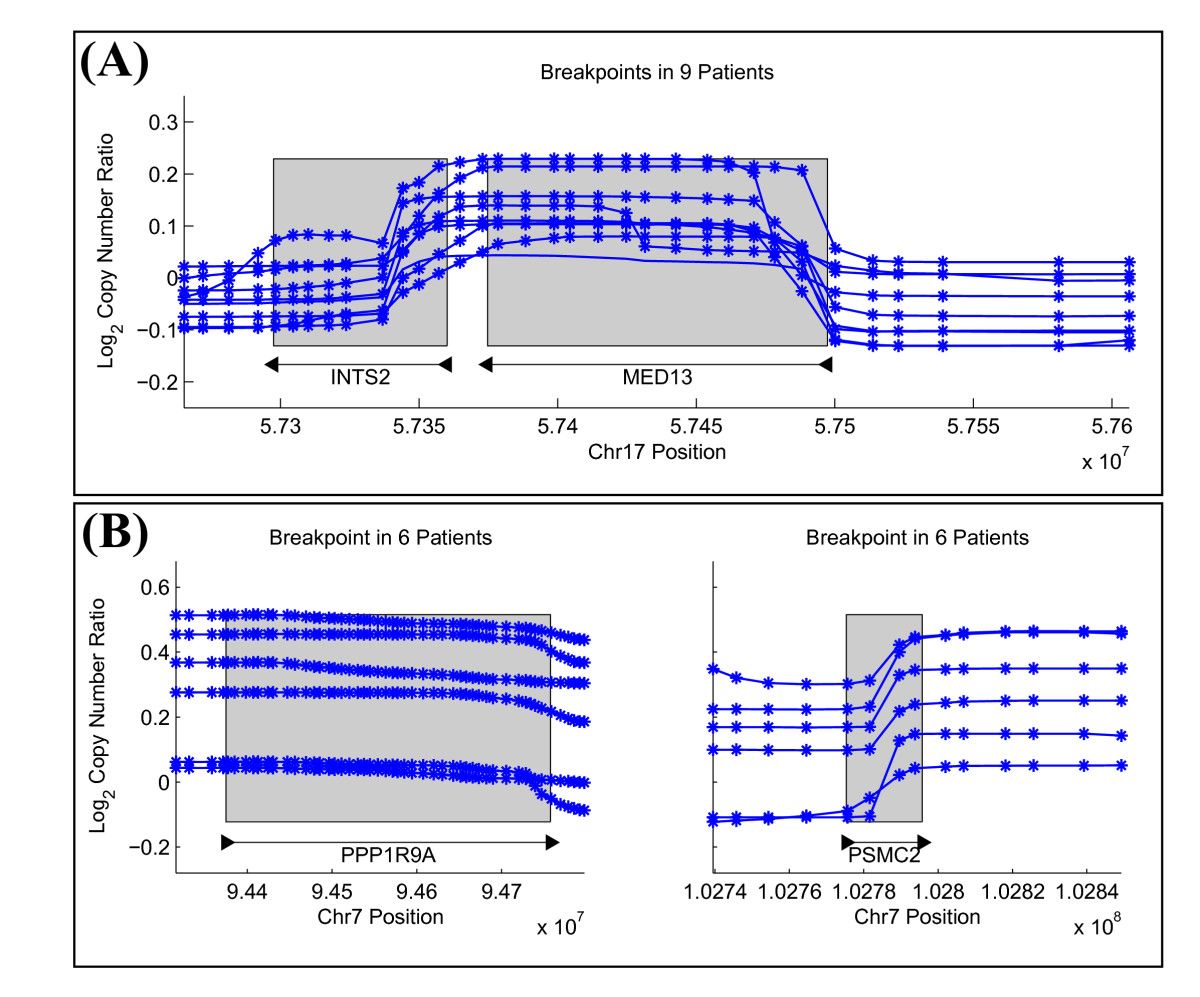
Structural variants (SVs) are large rearrangements of DNA that are an important contributor to genetic variation. During my PhD, I worked to identify SVs in human genomes from DNA microarray and sequencing technologies. To accommodate new third-generation sequencing platforms, we formalized a multi-linked read that generalizes the concept of paired reads. We developed two SV detection methods from multi-linked reads based on the sequencing platforms developed by Pacific Biosciences. SVs are also associated with a host of cancers, where somatic SVs - variants that are acquired within an individual's lifetime - are important for determining putative driver mutations. In the context of cancer biology, we developed an algorithm to identify recurrent fusion genes, where an SV combines two genes into a single, "hybrid" gene.