 Langmead B, Trapnell C, Pop M, Salzberg SL.
Langmead B, Trapnell C, Pop M, Salzberg SL. Renn Lab Thesis Project
2010 Reed Graduate
DAN BERNSTEIN
Determining the Accuracy of Expression Profiles Generated by Aligning RNA-Seq Data Against Heterospecific Reference Genomes
RNA-Seq is an emerging technology that has the potential to vastly exceed current
gene expression technologies (Marioni et al., 2008; Fu et al., 2009; Wang et al. 2009). Due to the fact that RNA-Seq is relatively new, as are
the multitude of bioinformatics programs that have been and are being developed, to
mine this high-throughput expression information, little research has been done consisting
of qualitative comparisons between and among these tools. In fact, according
to the literature searched over the course of this Thesis, there have been no qualitative
comparisons of expression profile accuracy for any RNA-Seq bioinformatics programs
other than those contained within the papers publish upon the release of a new program.
Those comparisons, however, almost entirely deal only with comparisons of
each program’s technical efficiency (i.e. speed of alignment, computer memory usage,
etc. . . ), and not with the biological accuracy. To my knowledge, this Thesis represents
the first (hopefully of many) qualitative comparison of the biological expression
profile results produced by these RNA-Seq bioinformatics programs.
In order to mimic the situation faced by researchers using non-model organisms for
RNA-Seq, I aimed to identify RNA-Seq datasets of a species for which full genome
sequences were available in multiple related species. The goal was to select different
related reference genomes representing different phylogenetic distances. The primate
lineage offers such datasets, however, the experimental design
is reversed from that which would be experience for most researchers
in ecologically relevant areas. Here, I
used a “model” RNA-Seq dataset (human) and “non-model” (other primate) reference genomes.
The expression dataset used in this thesiswas generated from Illumina sequencing of Homo sapiens kidney and liver samples (Human, NCBI SRA Accession: SRX000605”) [Marioni et al., 2008], and was obtained from NCBI’s Sequence Read Archive. In its entirety, the expression dataset contains 66,404,506 expression reads.
Six reference genomes wereused to generate the experimental
expression profiles: Pan troglodytes - Chimpanzee, NCBI Genome DB: “txid9598[Organism:noexp]”
Gorilla gorilla - Gorilla, NCBI Nucleotide Database: “FN568587:FN582117[PACC]”
Pongo ableii - Orangutan, NCBI Nucleotide
Database: “CM000550:CM000573[PACC]”
Macaca mulatta - Rhesus macaque,
NCBI Genome DB: “txid9544[Organism:noexp]”
Human - NCBI Nucleotide Database: “CM-
000663:CM000686[PACC]” (positive cont.)
Mus musculus - Mouse, NCBI Genome Database: “NC 000067:-
NC 000087[PACC]”
BOWTIE (v0.12.0, http://bowtiebio.sourceforge.net/index.shtml) and SHRiMP (v1.3.2, http://compbio.cs.toronto.edu/shrimp/) were installed on the Renn Lab student computer (Mac OSX v10.5.8, Processor 3.06 GHz Intel Core 2 Duo, Memory 4 GB 1067 MHz DDR3). Although SHRiMP was downloaded and installed on the
Renn Lab computer, due to slower-than-expected alignment speeds, a within-program
comparison of alignments using only BOWTIE was decided upon for the experimental
analysis of this thesis rather than a between-program comparison of alignments from
BOWTIE and SHRiMP.
The parameter value determining allowance of a certain number of mismatches was the basis for a qualitative comparison of these expression profile results. A true-positive set of expression reads was acquired from the conspecific alignments and an analysis of which heterospecific alignments recovered this set of true-positive reads was performed using BOWTIE and custom Perl scripts.
Results indicate that heterospecific alignments using the reference genomes from Chimpanzee and Gorilla are capable of returning the set of true-positive reads when the number of mismatches allowed is relaxed to at most 2 mismatches per read. These results suggest that it is possible for a heterospecific reference genome to be used when aligning RNA-Seq expression data given a certain species-to-species divergence time frame or species to-species genetic similarity.
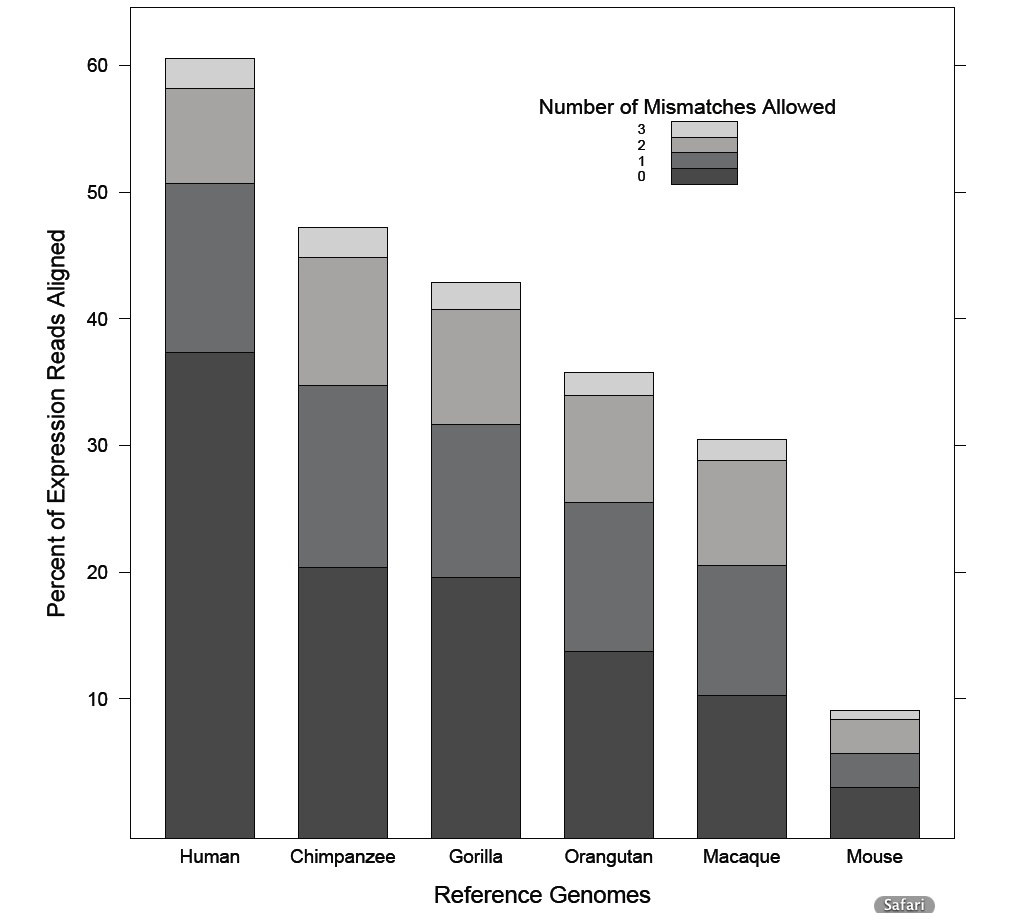 Figure1: Bar chart showing percentage of Human RNA-Seq expression reads that
Figure1: Bar chart showing percentage of Human RNA-Seq expression reads that
aligned to each reference genome for each mismatch allowance level. Percentages
are overlayed for all mismatches allowed. “Overlayed” means, for example, that the
bar for “3 Mismatches Allowed” using the Human Reference Genome is the total of
everything below (60.53%), not the width of the bar (2.32%). The difference in bar
heights shows how many more reads were found in “3 Mismatches Allowed” than “2
Mismatches Allowed” (2.32%). A number of data relationships can be observed in this
manifestation of the BOWTIE output. The first relationship is that the percentage of
the total number of reads returned decreases as the evolution divergence from Human
increases and the genomic identity to Human decreases. The second is that as the
mismatch allowance level is relaxed, the percentage of the total number of reads that is
returned increases, however, the increase in the percentage of reads returned presents
diminishing returns, meaning that the difference in percentage of reads returned at
one mismatch allowance level to the next decreases as the level increases.
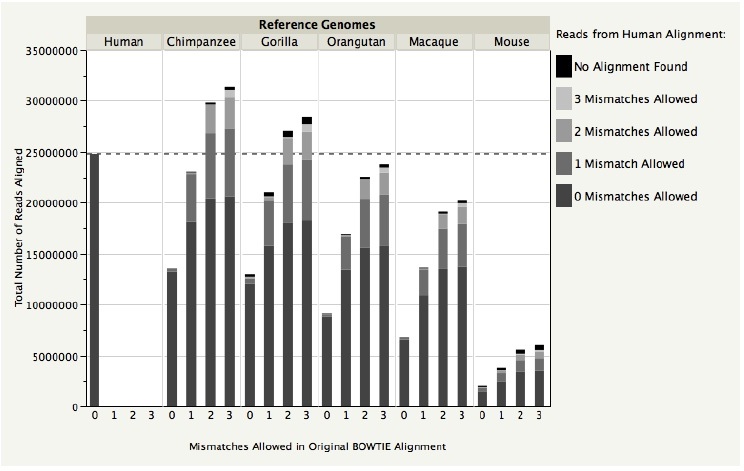 Figure 2: Stacked bar chart showing the total reads found for each heterospecific reference genome alignment and the composition of each alignment in terms of each Human-Mismatch read set. Chimpanzee (2 and 3 mismatches) and Gorilla (2 and 3 mismatches) were the only alignments that recovered a total number of reads greater than the number found in the true-positive BOWTIE alignment.
Figure 2: Stacked bar chart showing the total reads found for each heterospecific reference genome alignment and the composition of each alignment in terms of each Human-Mismatch read set. Chimpanzee (2 and 3 mismatches) and Gorilla (2 and 3 mismatches) were the only alignments that recovered a total number of reads greater than the number found in the true-positive BOWTIE alignment.
Work was done towards assigning these reads to biological identifiers (genes), however, the experiment was not performed. Those efforts included extensive perl scripting and command line perl script adapted from the scriptome (http://sysbio.harvard.edu/csb/resources/computational/scriptome/)
REFERENCES:
Fu, X., N. Fu, S. Guo, Z. Yan, Y. Xu, et al., 2009 Estimating accuracy of RNASeq
and microarrays with proteomics. BMC Genomics 10: 9.
Marioni, J. C., C. E. Mason, S. M. Mane, M. Stephens, and Y. Gilad, 2008
RNA-seq: An assessment of technical reproducibility and comparison with gene
expression arrays. Genome Research 18: 1509–1517
Wang, Z., M. Gerstein, and M. Snyder, 2009 RNA-Seq: a revolutionary tool
for transcriptomics. Nature Reviews Genetics 10: 57–63.