
Renn Lab Thesis Project
2011 Reed Graduate
Quinn Langdon
Hybe and go Seq: application of genomic tools to sociogenomic questions in the cichlid genus Julidochromis
It was my aim to compare the utility of RNA-Seq and microarray technology for application to sociogenomics. Specifically, I am interested in the social behavior of aggression related to sex-roles. I use the sex-role conventional J. transcriptus and sex-role reversed J. marlieri as models for differing sex-roles between species. These species have previously been studied at the behavioral level and also at the gene expression level (see Zero 2007, Schumer 2009, Wood 2010). To examine the molecular changes that accompany this heritable, yet plastic phenotype, I have used microarray techniques to determine relative gene expression within and between sexes and species. The novelty of this thesis lies in the direct comparison between species where all past work has focused on differences within a species. Therefore, where past analysis could only use gene lists to compare between species I have been able to do a more rigorous statistical analysis. I apply cutting edge microarray analyses in order to identify modules of coordinately expressed genes related to sex-role behavior and address the extent to which these patterns are similar or different between species. The technical obstacle that arises for direct comparison between species is that one must first verify that genomic DNA sequence differences are not confounding the expression difference that one aims to measure with microarray analysis. A concern with our heterologous hybridization technique is that underlying genomic sequence difference could bias the results.
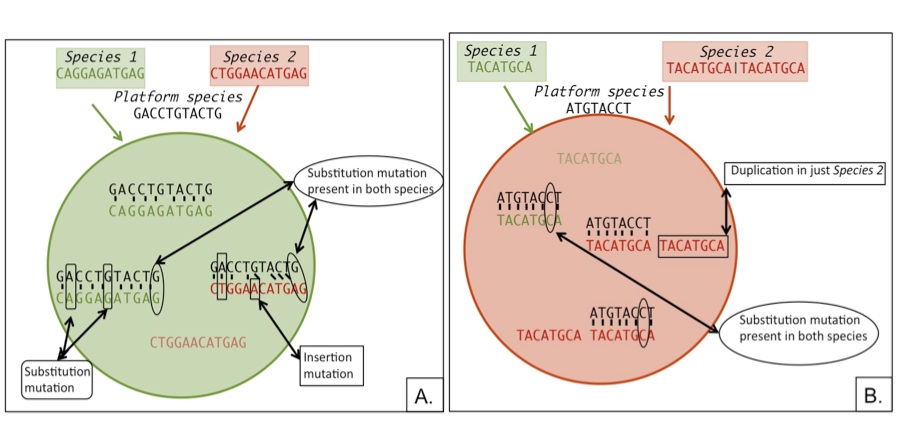 |
| Sequence difference bias on microarrays Cartoon showing how sequence differences will bias aCGH and expression microarray results. A. Shows how an insertion mutation will decrease species 2 binding affinity thereby making the feature appear biased towards species 1. B. Shows how a gene duplication in species 2 will increase the sequence’s abundance biasing the feature towards species 2. |
In order to identify the extent to which genomic sequence divergence would confound the gene expression results I performed competitive genomic hybridization using gDNA from the two Julidochromis species. In combination with the within and between species expression hybridizations, I then applied GenMask, a script written my Heather Machado (Renn et al 2009).
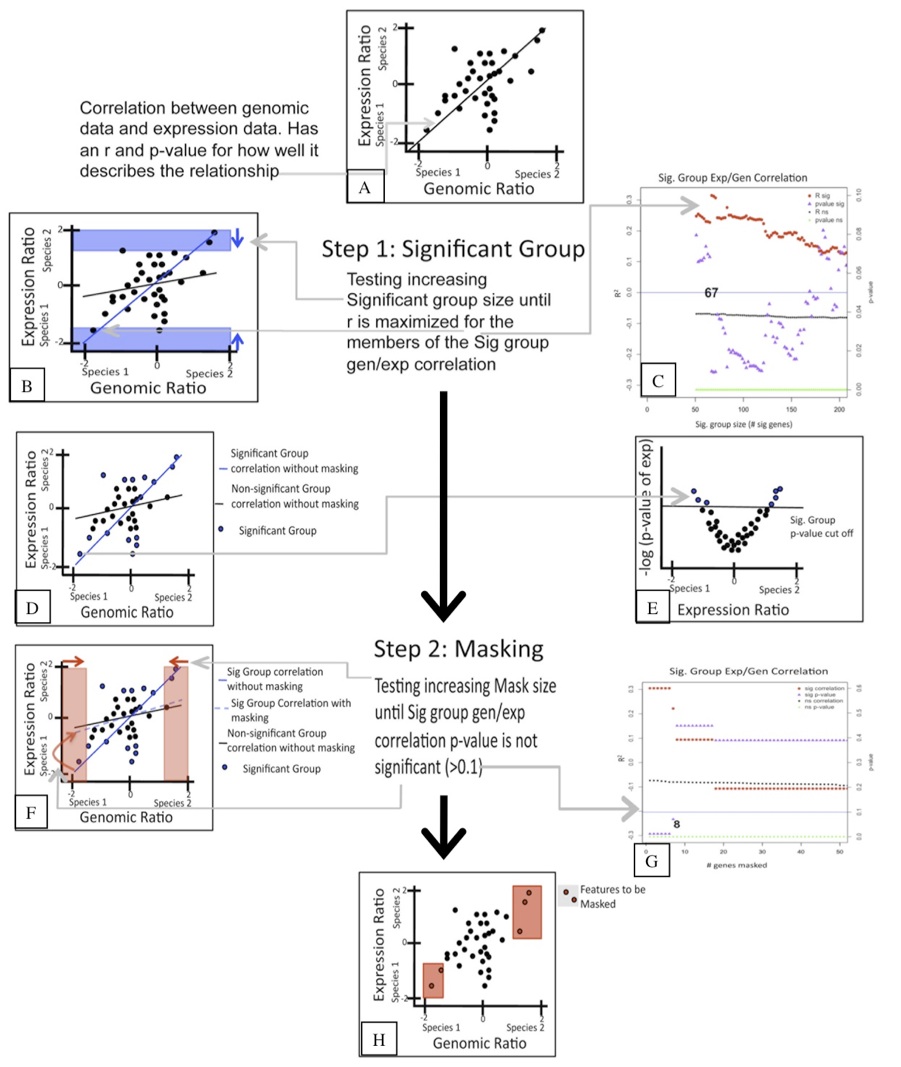
That analysis revealed that only 8 genes needed to be masked. Interestingly, many of those genes had previously been identified as varying in copy number between other cichlid species.
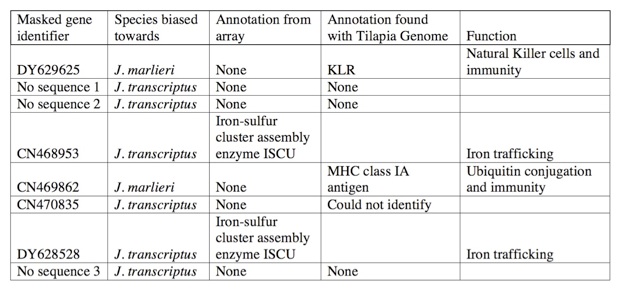
With the microarray mask in place, I could confidently analyze the between species expression analysis and identify coordinately regulated genes.
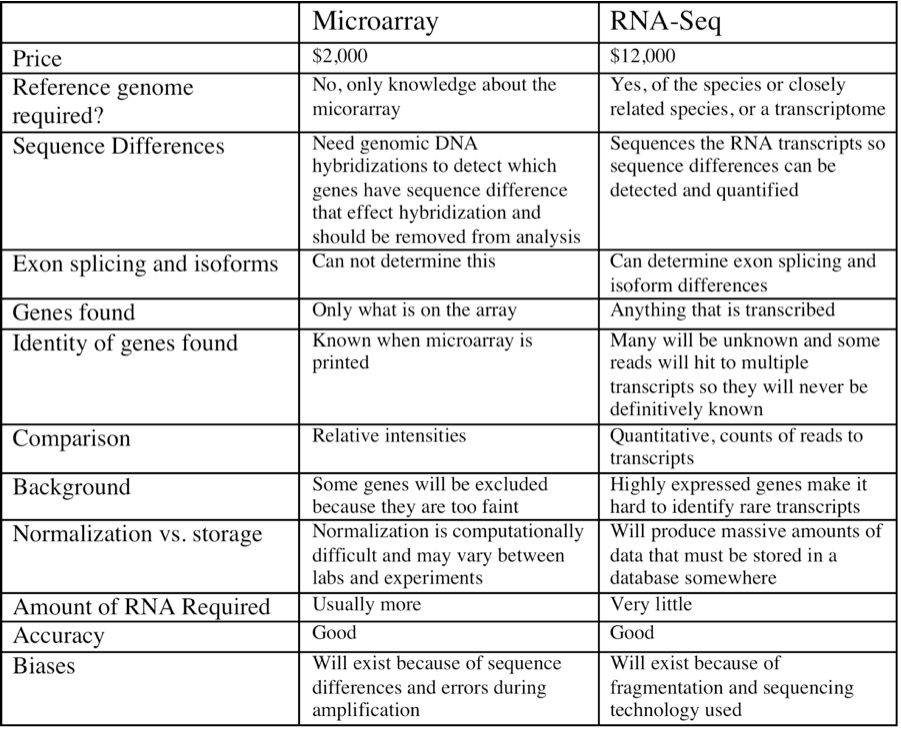
As technology advances, sequencing techniques are poised to replace microarrays. However, additional challenges face researchers who wish to use these tools in non-model species. RNA-Seq’s application to this project were discussed in detal.